Genomics Tool Development
Developing/evaluating genomic technologies and bioinformatic tools is an essential part of genomics research. As a part of our ongoing research effort, we develop computational tools, and evaluating technologies for large-scale genomic/proteomic data analysis. Here are a few examples.
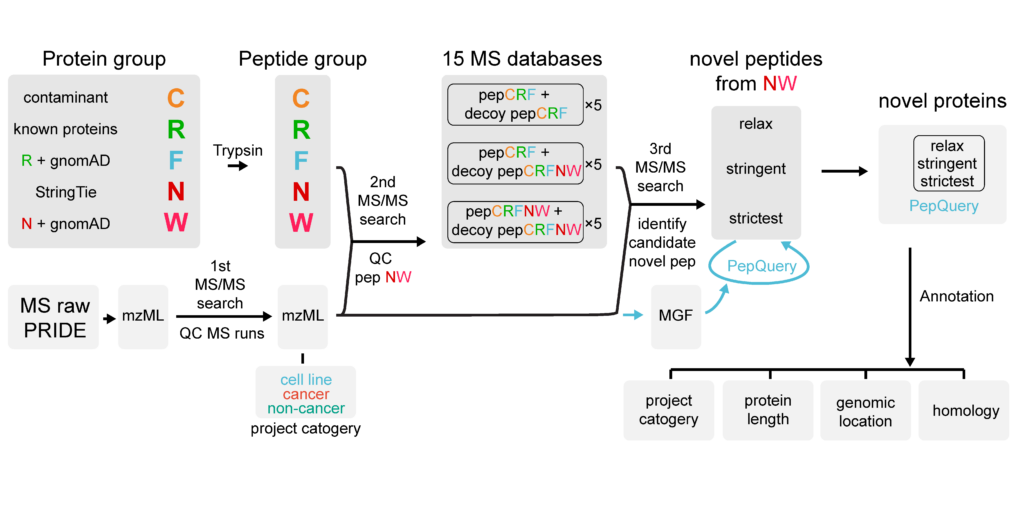
Identify novel proteins from mass-spec data using a proteogenomic approach
Although the human gene annotation has been continuously improved over the past 2 decades, numerous studies demonstrated the existence of a “dark proteome”, consisting of proteins that were critical for biological processes but not included in widely used gene catalogs. To provide a resource of high-confidence novel proteins from the dark proteome, we screened 50,000 mass spectrometry runs from over 900 projects to identify proteins translated from the Genotype-Tissue Expression transcript model with proteomic support. We also integrated 3.8 million common genetic variants from the gnomAD database to improve peptide identification. As a result, we identified 170,529 novel peptides with proteomic evidence, of which 6048 passed the strictest standard we defined.
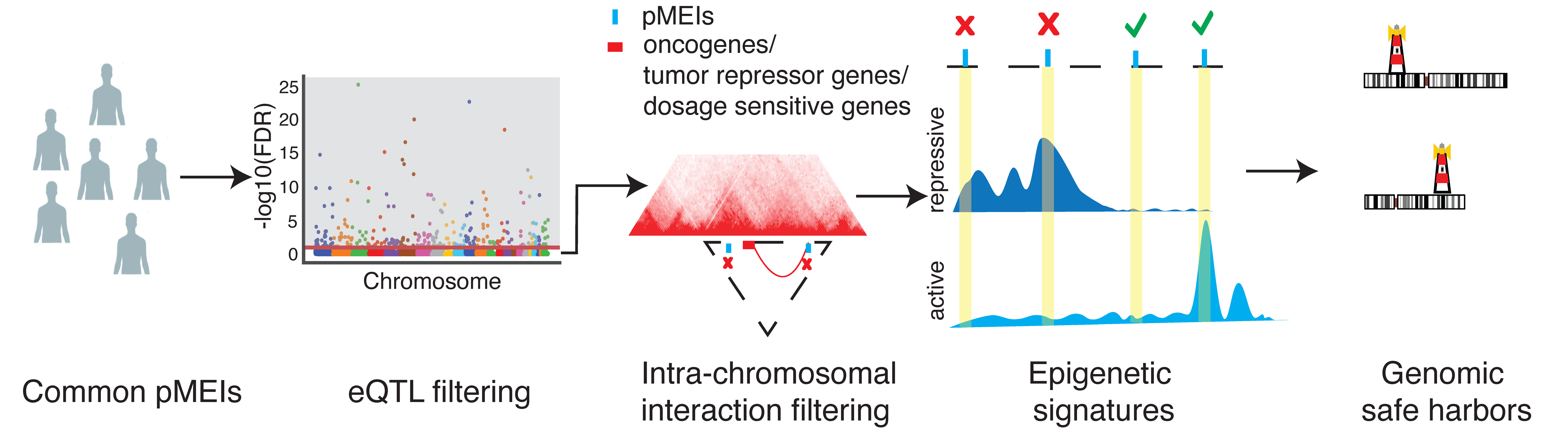
Identify genomic safe-harbor for gene therapy
Genomic safe harbors are regions of the genome that can maintain transgene expression without disrupting the function of host cells. Genomic safe harbors play an increasingly important role in improving the efficiency and safety of genome engineering. However, limited safe harbors have been identified. We develop a framework to facilitate searches for genomic safe harbors by integrating information from polymorphic mobile element insertions that naturally occur in human populations, epigenomic signatures, and 3D chromatin organization. By applying our framework to polymorphic mobile element insertions identified in the 1000 Genomes project and the Genotype-Tissue Expression (GTEx) project, we identify 19 candidate safe harbors in blood cells and 5 in brain cells. For three candidate sites in blood, we demonstrate the stable expression of transgene without disrupting nearby genes in host erythroid cells. We also develop a computer program, Genomics and Epigenetic Guided Safe Harbor mapper (GEG-SH mapper), for knowledge-based tissue-specific genomic safe harbor selection.
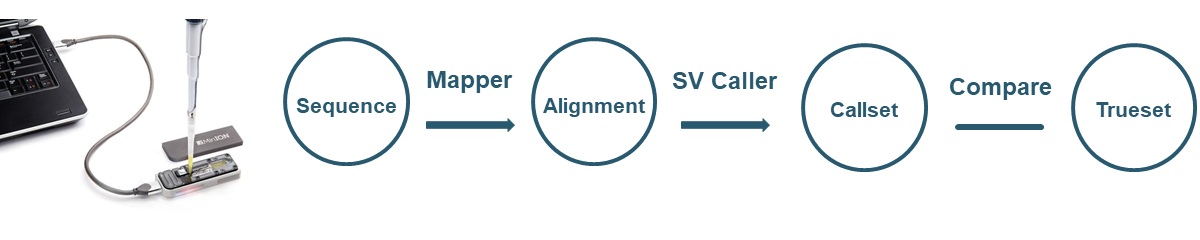
Evaluate different methods for structural variation identification using Nanopore sequencing
Structural variations (SVs) account for about 1% of the differences among human genomes and play a significant role in phenotypic variation and disease susceptibility. The emerging nanopore sequencing technology can generate long sequence reads and can potentially provide accurate SV identification. However, the tools for aligning long-read data and detecting SVs have not been thoroughly evaluated. Using four nanopore datasets, including both empirical and simulated reads, we evaluate four alignment tools and three SV detection tools. We also evaluate the impact of sequencing depth on SV detection. Finally, we develop a machine learning approach to integrate call sets from multiple pipelines.